前言
★ 文章导览★
滑动查看
AK-OTOF 非临床研究设计
1.1
结构及作用机制
2022年9月13日,美国 FDA 的一纸批文点燃了全球耳科医学界的希望——生物制药公司 Akouos 宣布,其基因治疗产品 AK-OTOF 获准进入临床试验。这不仅意味着全球首个针对遗传性耳聋的基因疗法正式迈向临床,更标志着人类首次通过 AAV 载体直接修复内耳功能,为 20,000 名因 OTOF 基因突变致聋的患者推开了一扇“听见世界”的大门。AK-OTOF 是一款双载体 AAVAnc80 药物,2022年10月,礼来公司斥资约 6.1 亿美元收购了 Akouos,以加速其听力基因疗法的开发进程。AK-OTOF 产品利用了 AAVAnc80 这种对耳蜗毛细胞(IHCs)具有高转导效率的衣壳,分别包装近6kb的人类 cDNA,并递送至内毛细胞中,以替代突变的基因,从而恢复内耳细胞的功能[3]。
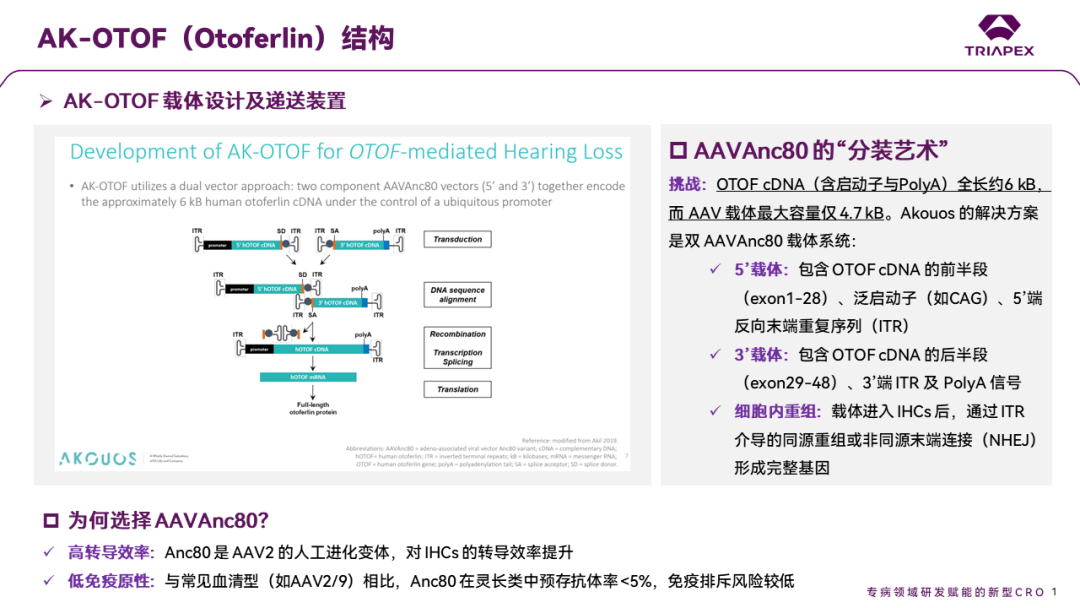
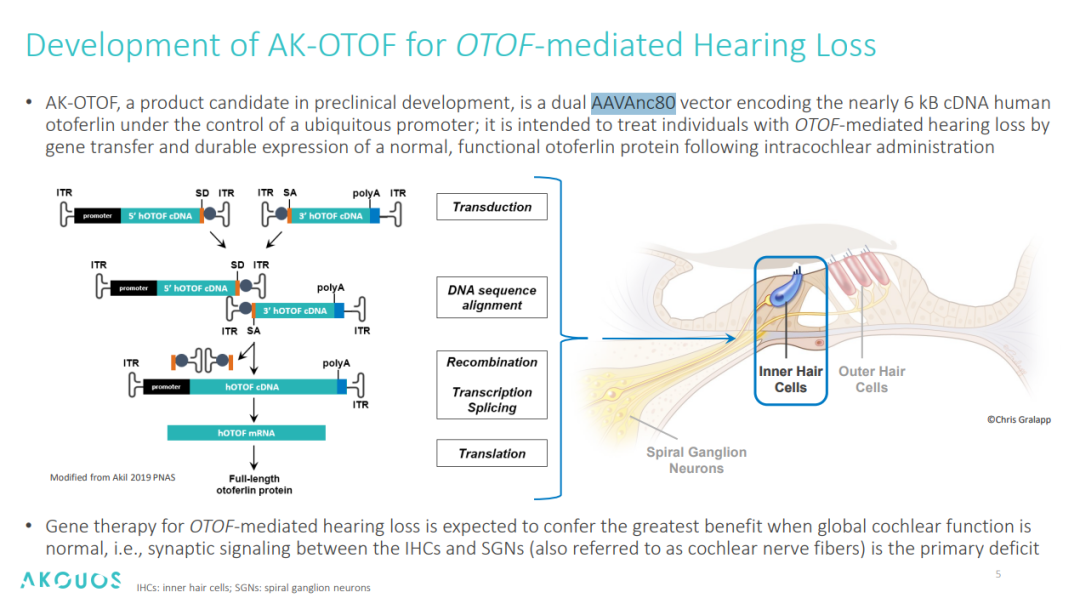
图2. AK-OTOF 作用机制和设计思路[3]
1.2
非临床研究设计及部分研究结果
2024年1月23日,Akouos 宣布了 1/2 期 AK-OTOF 研究的积极初步临床结果,其临床试验快速推进,取得积极成果的背后是非临床研究的严谨验证与临床策略的大胆创新。接下来,本文将聚焦非临床研究的关键数据与儿科临床试验的设计逻辑,深度解析这一突破性疗法如何从动物模型走向人类患者,为 OTOF 突变致聋的儿童重燃“听见世界”的希望。
1.2.1 非临床药效学研究
试验设计:
首先通过 CRISPR-Cas9 技术敲除小鼠 Otof 基因 exon7,构建 Otof 基因敲除小鼠(Otof⁻/⁻)模型,模拟人类 OTOF 无义突变,导致耳蜗内毛细胞无法释放神经递质,表现为听觉脑干反应(ABR)完全缺失,致聋小鼠。随后通过单侧耳蜗注射给药,将 AK-OTOF 注入小鼠耳蜗,并设置生理盐水对照组,1×(基准剂量)、2.5×、5×剂量组,探索 AK-OTOF 的药效学效应及剂量-反应关系。
研究结果:
首先是功能蛋白表达方面,免疫荧光的结果显示,3周龄小鼠单侧耳蜗注射 AK-OTOF(5×剂量)后,70% IHCs 在1个月内表达全长人源Otoferlin,且表达持续≥6个月。
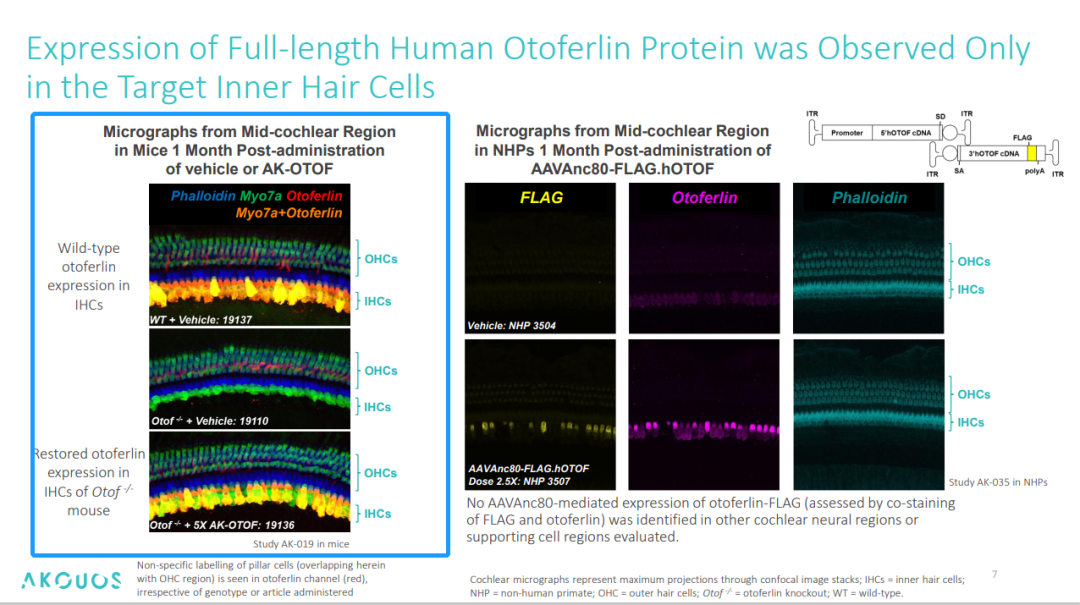
图3. 野生型小鼠与基因编辑小鼠 IHCs 细胞蛋白表达荧光图像[4]
在听觉功能恢复方面,AK-OTOF 在基因缺陷幼龄小鼠上也表现出了积极的结果。经 AK-OTOF 治疗的Otof⁻/⁻小鼠 ABR(听觉脑干反应)阈值从>100 dB SPL(Sound Pressure Level)恢复至 20-30 dB SPL(最快15天),这一数值与野生型小鼠相当。该结果意味着 AK-OTOF 重建了小鼠的听觉功能,且80%的小鼠在6个月后仍保持正常的ABR阈值,证实 AK-OTOF 基因表达的长期稳定性。同时,非临床数据显示5X剂量组疗效显著优于1X(P<0.01),也提示了一定的剂量反应关系。
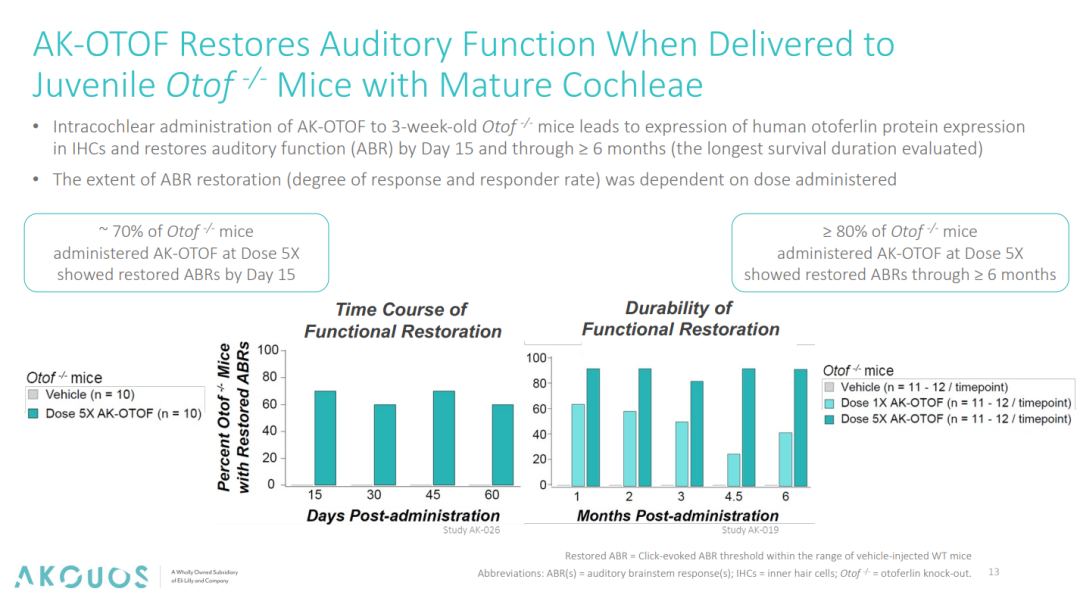
图4. AK-OTOF 恢复基因编辑小鼠听觉功能[4]
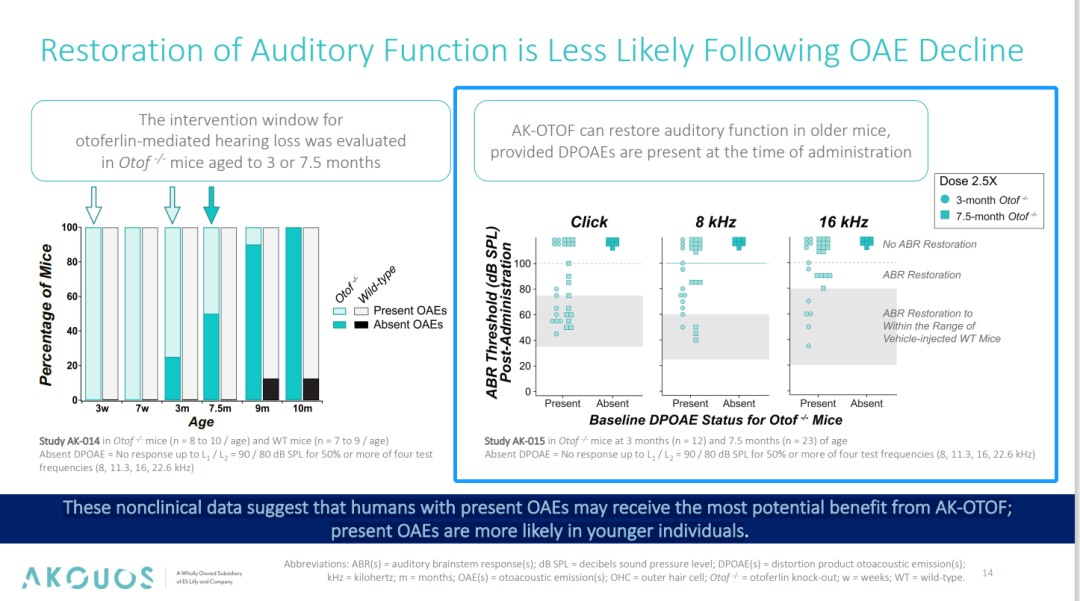
图5. AK-OTOF 对基因编辑小鼠 ABR 阈值的影响[4]
同时,我们注意到在耳声发射(OAEs:耳蜗内毛细胞功能的指标,Otof⁻/⁻小鼠在听力丧失前会经历 OAEs 的下降)下降之前,在Otof⁻/⁻小鼠耳蜗内给予 AK-OTOF,可恢复听觉功能,这一结果提示如果 AK-OTOF 在 OAEs 下降前(即听力损失的早期阶段)进行干预,可通过预防耳蜗细胞功能退化实现更有效的治疗,同时也可能为后续临床受试者人群的选择及治疗窗口的把握给出建议。
1.2.2 非临床安全性研究
图6. AK-OTOF 对食蟹猴听觉/耳蜗功能的影响[4]
图7. AK-OTOF 生物分布研究[4]

图8. AK-OTOF 病毒脱落研究[4]
安全性评价:在其它一般毒理评价指标中,耳蜗内给予 AK-OTOF 后,6 个月的毒性考察周期内没有观察到对食蟹猴及小鼠全身组织病理或临床病理、听觉的影响。

图9. AK-OTOF 非临床安全性研究结果[3]
综上,在食蟹猴和小鼠中,耳蜗内给药局部和全身耐受性良好,初步提示 AK-OTOF 具有较高的安全性。非临床研究所显示的“靶向性、持久性、安全性”构筑了 FDA 批准临床试验的科学基石。
AK-OTOF 临床研究布局
2022年9月,FDA 批准 Akouos 针对 AK-OTOF 提交的 IND 申请,允许开展首个针对遗传性听力损失的基因疗法临床试验,也是首个 AAV 载体直接递送至内耳的儿科试验。
2.1
Ⅰ/Ⅱ 期临床试验设计(NCT05821959)

图10. AK-OTOF 1/2期临床试验设计
AK-OTOF-101临床试验(NCT05821959)是一项1/2期试验,旨在评估通过 Akouos 递送装置给予递增剂量的AK-OTOF的安全性、耐受性和有效性。

图11. AK-OTOF 1/2期临床试验主要入排标准

图12. AK-OTOF 1/2期临床试验临床终点
2.2
当前研究结果
2024年1月23日,礼来公司的全资子公司Akouos 宣布了 AK-OTOF-101 研究的1/2期初步临床结果。该研究表明,第一位参与者(具有10多年重度听力损失史的个体)在给药 AK-OTOF 后 30 天内恢复听力!该受试者是一名11岁儿童,在接受 AK-OTOF 治疗前患有先天性严重听力损失,单次给药后 30 天内,ABR 阈值从无法检测恢复至65-20分贝(正常范围为≤20分贝),部分频率达到正常水平,即患者获得了听力功能的改善和恢复,可在不依赖人工耳蜗的情况下感知声音并做出反应;同时,在安全性方面,未报告严重不良事件,手术及药物耐受性良好。
据报道,Akouos 可能在耳鼻喉科研究协会(ARO)2024 Presidential Symposium 或 2025 MidWinter Meeting 会议公布第2例患者的数据。若后续试验验证了 AK-OTOF 的安全性和有效性,可能扩展至其他内耳疾病治疗,遗憾的是,笔者暂未查询到 AK-OTOF 第2例患者的临床数据,但是2025年2月25日,再生元(Regeneron)公布了其基因疗法 DB-OTO 的1/2期数据,DB-OTO 也是针对遗传性耳聋开发的一款双载体 AAV 基因治疗产品,由再生元于收购其长期开发合作伙伴 Decibel Therapeutics 所得,与 AK-OTOF 的作用机制类似,DB-OTO 也通过将 OTOF cDNA 分段包装于两个 AAV 载体中,以解决因 OTOF 基因过大而超出单个 AAV 载体载荷的问题。
再生元公布的数据显示,在1/2期试验中,12 名儿童接受了 DB-OTO 治疗,其中 9 名患者的接受了单侧 DB-OTO 耳蜗注射的治疗,另外 3 名患者(约 1 岁)则接受了双侧耳蜗注射的治疗,本次更新的内容主要包括给药后 72 周观察的结果,具体如下:
当前 AK-OTOF 及 DB-OTO 的积极性结果为全球约 20 万 OTOF 突变患者提供了一次性治愈可能,有望帮助他们摆脱传统的助听器或人工耳蜗,也能够推动基因疗法在感觉神经系统疾病中的应用,为其他遗传性耳聋类型(如 GJB2 突变)提供技术参考。但仍应注意到该类产品的长期安全性和技术复杂性,双载体递送和精准耳蜗给药对手术操作要求极高,也需要持续监测基因表达稳定性和免疫反应等风险。总之,创新的双 AAV 载体递送和精准耳蜗给药技术,展现了基因疗法在遗传性耳聋治疗中的潜力。早期临床数据显示其安全有效,未来需进一步验证长期疗效及扩大适应症。
AK-OTOF 非临床研究的临床转化价值
通过以上非临床及临床研发路径的梳理和总结,总体来说,非临床研究为临床研究提供了疗效验证、安全性保障、剂量选择等多方面的支持,AK-OTOF 临床策略的大胆创新,离不开非临床研究数据的缜密性与完整性,为 AK-OTOF 的临床研究提供了全方位科学支持。
3.1
疗效验证与机制支持
靶向性验证
非临床研究通过Otof⁻/⁻小鼠和灵长类(NHPs)模型证实,AK-OTOF 能够特异性表达于IHCs,未在外毛细胞、螺旋神经节或其他非靶组织中检测到功能性蛋白。这提示临床研究可聚焦 IHCs 功能恢复作为核心疗效指标。
持久性证据
在药效学研究中,小鼠中单次给药后疗效持续≥6个月,且食蟹猴生物分布数据也提示耳蜗内载体 DNA 长期存在,支持短周期的临床研究设计单次给药方案。
3.2
安全性保障
局部耐受性
在毒理学研究中,食蟹猴中耳蜗毛细胞存活率>98%,无炎症或纤维化,证实手术递送方式的安全性,为临床选择微创经外耳道入路提供依据。
系统安全性
NHPs及小鼠中中血液生化、肝肾功能、脑组织病理均无异常,且生物分布数据提示非靶组织(肝脏、脾脏)中AAV载体快速清除,提供了疗效和安全性数据,可能减少临床研究中对全身毒性的担忧。
3.3
剂量选择与优化
剂量反应曲线
小鼠实验中,5X剂量组疗效显著优于1X(P<0.01),但高于5X剂量组后疗效是否会出现平台期还未尝可知,提示临床需探索最佳剂量区间(如2.5X-5X),避免无效或过度治疗。
剂量换算逻辑
基于内耳体积(人类耳蜗≈80 μL,小鼠≈1 μL)进行跨种属剂量换算,保留 10 倍安全边际,为临床起始剂量提供科学依据。
3.4
干预窗口与人群选择
OAE 动态变化
动物数据显示,OAE 存在时(小鼠<3月龄,人类<11岁)治疗可最大化疗效,支持临床入组OAE 阳性儿童(2-11岁),锁定最佳治疗干预窗口。
基因型-表型关联
非临床研究聚焦双等位 OTOF 致病突变,提示临床需严格筛选突变患者,确保目标人群同质性。
FDA 的“绿色通道”—— AK-OTOF 孤儿药认定与儿科试验的监管逻辑
通常,监管机构如 FDA 在审批临床试验申请时,一般要求先在成人中开展临床试验,积累过一定的人体数据之后,再逐步外推到儿童,尤其是对于新药。但对于 AK-OTOF 来说,FDA 为何会支持直接在儿童群体中开展临床研究?其背后不仅体现了 Akouos 对产品开发科学性的严格把控,更离不开对监管策略的灵活运用,以加速其儿科适应症的开发。
4.1
孤儿药认定与罕见儿科病的双重加持
孤儿药认定(ODD):基于 OTOF 耳聋在美国的患病率<20万,AK-OTOF 获得孤儿药认证,Akouos 获得 7 年市场独占权、50%临床试验税收抵免及 FDA 全程指导。
罕见儿科病(RPDD):允许在无成人数据时优先开展儿科试验,尤其适用于无替代疗法的严重疾病。AK-OTOF 于2021年斩获两项认定(早于 IND 批准时间),为儿科试验铺平道路。
4.2
为何跳过成人直接入组儿童?
科学依据
监管策略
综上所述,该案例体现了 FDA 对罕见病治疗创新的支持,尤其在儿科领域优先考虑患者群体需求与治疗窗口期的特殊性。AK-OTOF 能够在儿童中直接开展临床试验的原因主要有以下几点:
听见未来 —— 基因疗法的希望之光
AK-OTOF 的非临床研究以“靶向性、持久性、安全性”三大支柱,构筑 了 FDA 放行儿科试验的科学基石;而临床试验的精心设计,则体现了对儿科罕见病患者需求的深刻回应。从 OTOF 基因的碱基对到 FDA 的审评会议,从波士顿实验室的显微操作到非洲村庄的听力筛查,每一步都写满了创新者的执着与患者家庭的期盼。正如 Akouos 创始人 Manny Simons 所言:“我们修复的不只是听力,而是重新连接一个人与世界的对话。”或许在未来某天,基因疗法能让每一个“寂静星球”的孩子,听见花开的声音。
鼎泰集团耳科药物非临床评价平台技术能力
图13. 鼎泰集团耳科药物非临床评价平台
转化科学与药政策略部
毒理II部耳科药物评价团队
