万事开头难,看好文章就不难:ITSxpress: Software to rapidly trim internally… | F1000Research
仓库地址:GitHub – USDA-ARS-GBRU/q2_itsxpress: A plugin for qiime2 that runs ITSxpress
还有这个EOL分支版:GitHub – USDA-ARS-GBRU/itsxpress at 1.8.1-EOL
这个插件就是作者将 ITSx – Microbiology.se 这个软件做成了qiime2的插件,因此,大家可以先了解一下ITSEXPRESS 和 ITSx。
GitHub – USDA-ARS-GBRU/itsxpress: Software to trim the ITS region of FASTQ sequences for amplicon sequencing analysis
ITSx – Microbiology.se , GitHub – alk224/akutils-v1.1.1: Mostly useful scripts and files for processing MiSeq amplicon data
akutils这个仓库比较老了,但还有很多东西是可以借鉴一下的。下面这个是新版也好几年了。
GitHub – alk224/akutils-v1.2: akutils with massive updates over v1.1 to functionality and command line interface.
itsx的文章也要看:
https://besjournals.onlinelibrary.wiley.com/doi/10.1111/2041-210X.12073
qiime的最方便方式还是在conda安装了,所以这里也是讲现成conda环境下已安装qiime2的情况下进行安装配置。
github里面讲的是使用mamba加速器,但如果是服务器集群还是建议大家测试完成后再使用mamba命令,因为解释器切换,有可能造成安装源配置错误的问题,部分软件版本不兼容,后期安装和配置新环境的时候可能引起不少莫名其妙的错误,所以如果现在为conda解释器的不要急着换成mamba,做好测试再切换。所以这里还是使用conda
## 激活原有环境
source activate qiime2-202309
## 按照官网直接安装1.8.1可能会报错,现状express,默认最新版
conda install -c bioconda itsxpress
#我这里装完后是这样:
added / updated specs:
- itsxpress
The following packages will be downloaded:
package | build
---------------------------|-----------------
bbmap-39.01 | h92535d8_1 9.0 MB bioconda
biopython-1.78 | py38h7f8727e_0 2.2 MB
certifi-2023.11.17 | py38h06a4308_0 158 KB
itsxpress-1.8.0 | py_1 968 KB bioconda
------------------------------------------------------------
Total: 12.2 MB
The following NEW packages will be INSTALLED:
bbmap bioconda/linux-64::bbmap-39.01-h92535d8_1
biopython pkgs/main/linux-64::biopython-1.78-py38h7f8727e_0
itsxpress bioconda/noarch::itsxpress-1.8.0-py_1
The following packages will be UPDATED:
ca-certificates conda-forge::ca-certificates-2023.7.2~ --> pkgs/main::ca-certificates-2023.08.22-h06a4308_0
certifi conda-forge/noarch::certifi-2023.7.22~ --> pkgs/main/linux-64::certifi-2023.11.17-py38h06a4308_0
The following packages will be SUPERSEDED by a higher-priority channel:
openssl conda-forge::openssl-1.1.1w-hd590300_0 --> pkgs/main::openssl-1.1.1w-h7f8727e_0
###################### 也就是1.8.0版,但后面安装q2插件后会升到了1.8.1版 ######################### 继续安装q2 插件
pip install q2-itsxpress
####################
Installing collected packages: pyzstd, biopython, itsxpress, q2-itsxpress
Attempting uninstall: biopython
Found existing installation: biopython 1.78
Uninstalling biopython-1.78:
Successfully uninstalled biopython-1.78
Attempting uninstall: itsxpress
Found existing installation: itsxpress 1.8.0
Uninstalling itsxpress-1.8.0:
Successfully uninstalled itsxpress-1.8.0
Successfully installed biopython-1.81 itsxpress-1.8.1 pyzstd-0.15.9 q2-itsxpress-1.8.1
qiime dev refresh-cache### 官网命令:
qiime itsxpress
###############################
Error: QIIME 2 has no plugin/command named 'itsxpress'. Did you mean 'q2-itsxpress'?
### 哈哈,报错了。
### 按照提示使用q2 插件。
qiime q2-itsxpress
# 帮助信息出来了,应该没问题了。
Usage: qiime q2-itsxpress [OPTIONS] COMMAND [ARGS]...
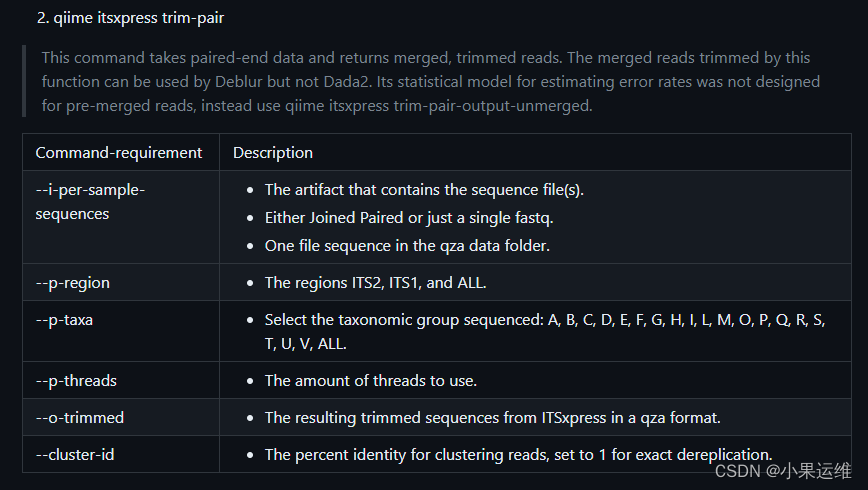
Description: #####This is the end of life version 1 of ITSxpress. Please
check Github for version 2of ITSxpress.#####ITSxpress trims amplicon reads
down to their ITS region. ITSxpress is designed to support the calling of
exact sequence variants rather than OTUs. This newer method of sequence
error-correction requires quality score data from each sequence, so each
input sequence must be trimmed. ITSxpress makes this possible by taking
FASTQ data, de-replicating the sequences then identifying the start and stop
sites using HMMSearch. Results are parsed and the trimmed files are
returned. The ITS 1, ITS2 or the entire ITS region including the 5.8s
rRNAgene can be selected. ALL requires very long reads so it is not
routinelyused with Illumina data. ITSxpress uses the hmm models from ITSx so
results are comparable.
Plugin website: https://github.com/USDA-ARS-GBRU/q2_itsxpress
ITSxpress: https://github.com/USDA-ARS-GBRU/itsxpress
Getting user support: Please post to the QIIME 2 forum for help with this
plugin: https://forum.qiime2.org
Options:
--version Show the version and exit.
--example-data PATH Write example data and exit.
--citations Show citations and exit.
--help Show this message and exit.
Commands:
trim-pair Trim paired-end reads, output merged reads for
use with Deblur
trim-pair-output-unmerged Trim paired-end reads, output unmerged reads for
use with Dada2
trim-single Trim single-end reads
用例:使用两个cpu线程,使用PairedSequencesWithQualty qza从真菌扩增子测序数据集中修剪ITS2区域。使用的示例文件位于paired.qza下的Tests文件夹中。
qiime itsxpress trim-pair --i-per-sample-sequences ~/parired.qza --p-region ITS2
--p-taxa F --p-threads 2 --o-trimmed ~/Desktop/out.qza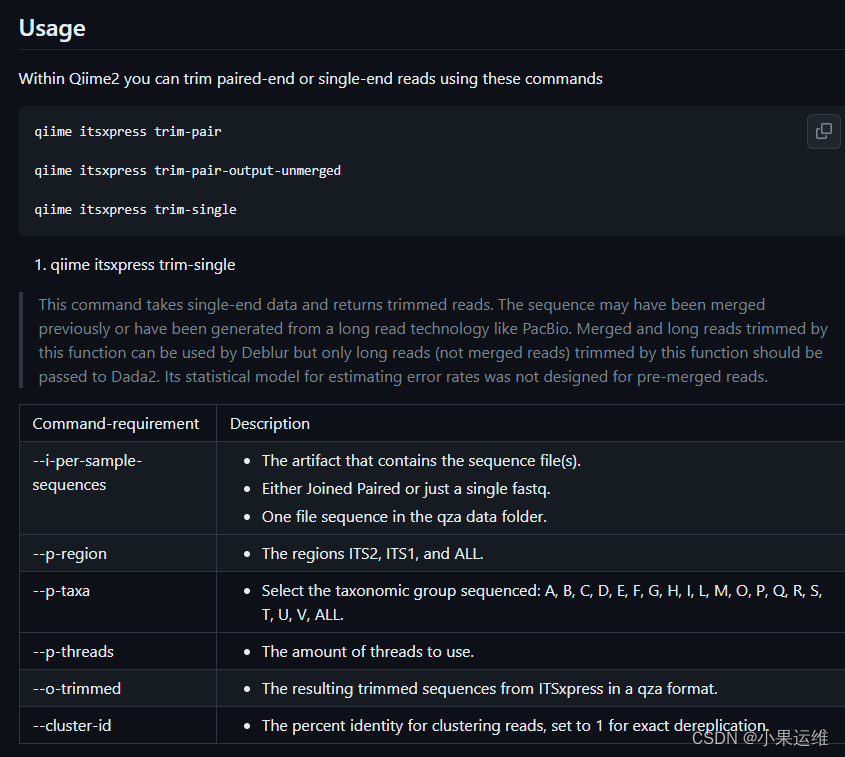

先单独建一个环境:
conda create -n ITSxpress_V1EOL python=3.8.13
conda activate ITSxpress_V1EOL再安装itsxpress
### conda 安装,有可能报错,建议吧==1.8.1去掉直接安装,会是1.8.0.
conda install -y -c bioconda itsxpress==1.8.1
### pip 安装,需要先安装依赖, pip安装的是1.8.1
conda install -y -c bioconda hmmer==3.1b2
conda install -y -c bioconda bbmap==38.69
conda install -y -c bioconda vsearch==2.21.1
pip install itsxpress
usage: itsxpress [-h] –fastq FASTQ [–single_end] [–fastq2 FASTQ2] –outfile OUTFILE [–outfile2 OUTFILE2] [–tempdir TEMPDIR] [–keeptemp] –region {ITS2,ITS1,ALL}
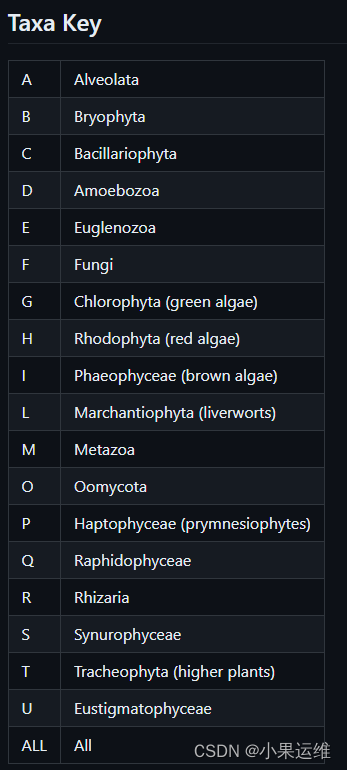
[–taxa {Alveolata,Bryophyta,Bacillariophyta,Amoebozoa,Euglenozoa,Fungi,Chlorophyta,Rhodophyta,Phaeophyceae,Marchantiophyta,Metazoa,Oomycota,Haptophyceae,Raphidophyceae, Rhizaria,Synurophyceae,Tracheophyta,Eustigmatophyceae,All}]
[–cluster_id CLUSTER_ID] [–reversed_primers] [–log LOG] [–threads THREADS]
itsxpress: error: the following arguments are required: –fastq/-f, –outfile/-o, –region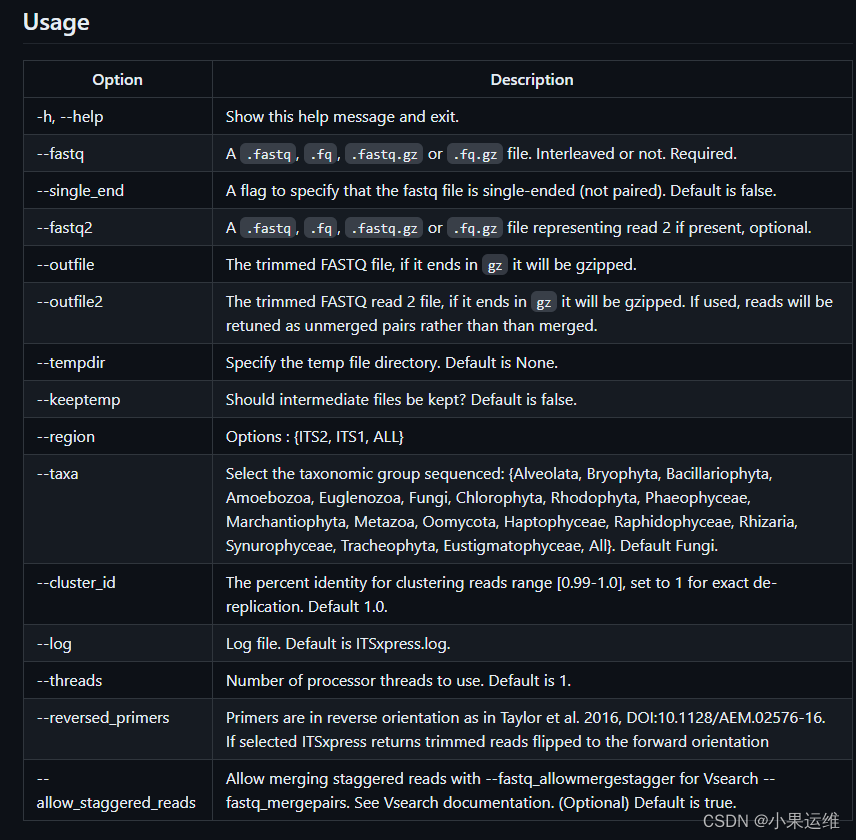
使用示例:
#Examples
#Use case 1: Trimming the ITS2 region from a fungal amplicon sequencing dataset with forward and reverse gzipped FASTQ files using two cpu threads. Return a single merged file for use in Deblur.
itsxpress --fastq r1.fastq.gz --fastq2 r2.fastq.gz --region ITS2
--taxa Fungi --log logfile.txt --outfile trimmed_reads.fastq.gz --threads 2
#ITSxpress can take gzipped or un-gzipped FASTQ files and it can write gzipped or un-gzipped FASTQ files. It expects FASTQ files to end in: .fq, .fastq, .fq.gz or fastq.gz.
#Use case 2: Trimming the ITS2 region from a fungal amplicon sequencing dataset with forward and reverse gzipped FASTQ files using two cpu threads. Return a forward and reverse read files for use in Dada2.
itsxpress --fastq r1.fastq.gz --fastq2 r2.fastq.gz --region ITS2
--taxa Fungi --log logfile.txt --outfile trimmed_reads.fastq.gz --threads 2
#ITSxpress can take gzipped or un-gzipped FASTQ files and it can write gzipped or un-gzipped FASTQ files. It expects FASTQ files to end in: .fq, .fastq, .fq.gz or fastq.gz.
#Use case 3: Trimming the ITS2 region from a fungal amplicon sequencing dataset with an interleaved gzipped FASTQ files using two cpu threads. Return a single merged file for use in Deblur.
itsxpress --fastq interleaved.fastq.gz --region ITS2 --taxa Fungi
--log logfile.txt --outfile trimmed_reads.fastq.gz --threads 2
# Use case 4: Trimming the ITS2 region from a fungal amplicon sequencing dataset with an single-ended gzipped FASTQ files using two cpu threads.
itsxpress --fastq single-end.fastq.gz --single_end --region ITS2 --taxa Fungi
--log logfile.txt --outfile trimmed_reads.fastq.gz --threads 2
# Single ended data is less common and may come from a dataset where the reads have already been merged.
# Use case 5: Trimming the ITS1 region from a Alveolata amplicon sequencing dataset with an interleaved gzipped FASTQ files using 8 cpu threads.
itsxpress --fastq interleaved.fastq.gz --region ITS1 --taxa Alveolata
--log logfile.txt --outfile trimmed_reads.fastq.gz --threads 8q2-itsxpress这个插件目前只兼容到1.8.1版,且pip版的已经停止维护和升级;要使用最新的2.0.1 ,需要再conda单独安装一次,且运行时直接使用命令即可。建议单独创建一个环境再安装
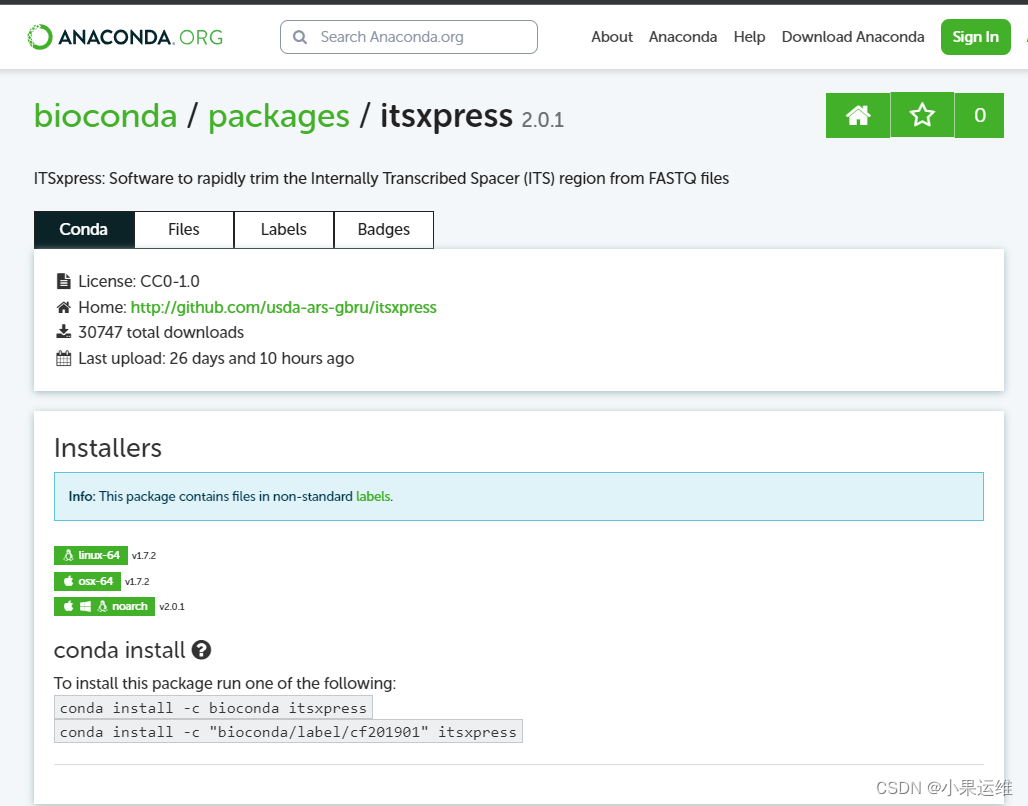
conda install -c bioconda itsxpress
