一、医疗器械注册与备案管理分类界定
1)按照《医疗器械注册与备案管理办法》的法律依据。第一类医疗器械实行产品备案管理,到企业所在市、或设区的药品监督管理局申请备案;
第二类、第三类医疗器械实行产品注册管理,分别到省和国家药品监督管理局注册申请;(关于进口第一类医疗器械备案,进口第二类、第三类医疗器械注册制度,均由国家药品监督管理局审查);
注:对新研制的尚未列入分类目录的医疗器械,申请人可以直接申请第三类医疗器械产品注册,也可以依据分类规则判断产品类别并向国家药品监督管理局申请类别确认后,申请产品注册或者进行产品备案。申请适用创新产品注册程序的,申请人应当在产品基本定型后,向国家药品监督管理局提出创新医疗器械审查申请。国家药品监督管理局组织专家进行审查,符合要求的,纳入创新产品注册程序。
2)按照《医疗器械经营监督管理办法》经营一类医疗器械不需要备案,二类医疗器械到市或设区备案登记,三类医疗器械申请经营许可证。
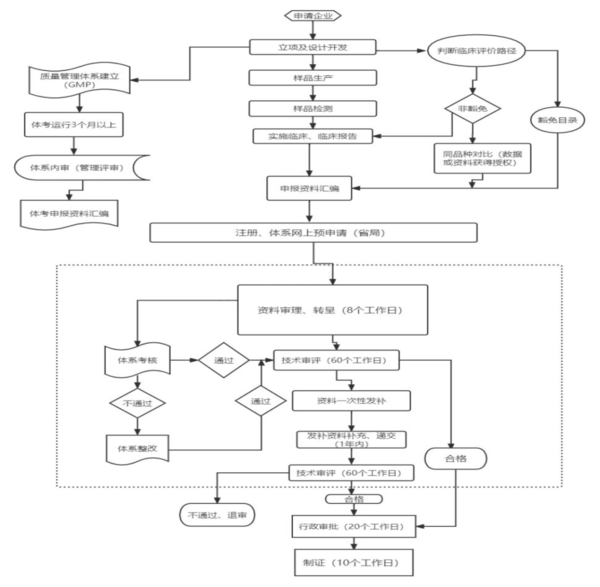
二、医疗器械注册申报流程
1. 申请企业对产品立项及设计开发
2.样品生产及检测送检
3.判断临床评价路径,可依据总局发布的免于临床评价医疗器械目录(2021年)判定产品是否属于免临床,如果免临床进行同产品对比(数据或资料获得授权);非豁免则实施临床及临床报告(临床细则见下文)
4.质量管理体系GMP建立且体系运行3个月以上,我司辅助企业进行管理评审
5.注册、体系网上预约申请(二类省局、三类国家局)
6.器械评审中心对申请资料审理
7.评审合格后20个工作日内行政审批,进行制证。
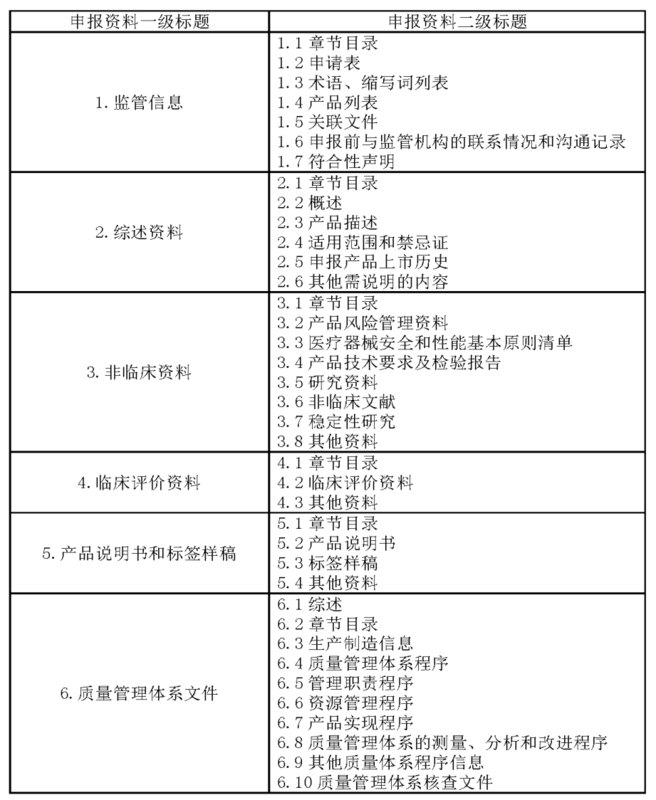
三、医疗器械注册申报资料要求
四、临床试验流程
1.选择CEO/CRC机构
2. 临床实验机构筛选
3. 临床试验方案确认
4. 机构备案
5. 伦理审查(获取伦理批件)
6. 临床实验备案
7. 临床项目启动
8. 受试者入组
9. 方案实施
10. 临床观察
11. 数据收集与监测: 在试验期间,对受试者的数据进行收集和监测,确保数据的准确性和完整性。同时,对试验过程中的安全性进行监测,及时发现和处理不良事件。
12. 数据分析与结果解释: 在试验结束后,对试验数据进行统计分析,评估试验产品的安全性和有效性。通过数据分析得出结论并解释试验结果。
13. 报告撰写与提交: 撰写试验报告,将试验结果提交给监管机构进行审批。报告应该包括试验的目的、方法、结果、结论和安全性等信息。
14. 随访和长期观察: 在试验结束后,可能需要进行长期的随访和观察,以进一步评估试验产品的长期安全性和效果。
15. 注册与发布: 在完成临床试验后,可以将试验结果进行注册,并根据适当的政策要求和科学伦理,在科学期刊上发布试验结果。
五、生产许可证申请:
1. 申请表
2. 营业执照
3. 人员信息
4.产地证明文件(场地证明的符合性:1、要提供房产证或场地使用证 2.或镇街道办盖章 3.场地使用性质:办厂、办公、生产)
4. 设备清单
5. 工艺流程图
6. 授权委托书
7.注册人制度:两个企业签订的协议、委托方和被委托方的资质文件、营业执照
注:关于医疗器械延伸检查,广东省药监给出政策解读!http://www.hxjiamei.com/h-nd-203.html
【华夏佳美】一站式跨境服务平台,专注医疗器械注册申请、GMP体系建立辅导、美国FDA510k、QSR820体系建立、欧盟CE认证、MDR、IVDR技术文件编写、ISO13485体系技术指导等提供全球医疗器械合规咨询服务,详情请咨询在线客服或官方电话:400-069-8010。
一、MoCRA法案实施背景
2023年8月7日美国食品和药品监督管理局即FDA根据《2022年化妆品监管现代化法案》(MoCRA)的规定,发布了关于化妆品工厂注册和产品清单的指南草案。 于2023 年 3 月 27 日结束了该自愿注册计划,并计划在2023 年 10 月 推出新的注册系统以满足法规规定的化妆品工厂注册和产品列名的要求。
二、谁需要向FDA 提供化妆品的注册?
1)设施注册:化妆品制造商和加工商必须向FDA注册设施
2)产品清单:负责人必须向FDA 列出每种上市的化妆品,包括产品成分
解释:负责 人是指根据《FD&C 法案》第 609(a) 条或《公平包装和标签法案》第 4(a) 条,其名称出现在化妆品标签上的化妆品制造商、包装商或分销商。
三、关于FDA化妆品注册的豁免
1.MoCRA 免除某些小型企业的设施注册和产品列名要求。
但是,此类豁免不适用于制造或加工以下化妆品的设施或负责人:
1)在习惯或通常使用条件下经常接触眼睛粘膜的产品。
2)注射的产品。
3)供内部使用的产品。
4)旨在在习惯或通常使用条件下改变外观超过24 小时并由消费者移除的产品不属于此类使用条件的一部分。
5)仅参与以下活动的企业:贴标(Labeling),重新贴标(Relabeling),包装(Packaging),重新包装(Repackaging),储存(Holding),分销(Distributing)。
2.这里的包装(Packaging)和重新包装(Repackaging)不包含把化妆品装进容器中的活动,参与此类活动的企业需要完成注册。
3.既是药品又是化妆品的产品,其必须按照药品完成注册,这时便无需按照化妆品注册。
四、关于FDA化妆品注册更新的周期
1)设施需要并每两年更新一次注册
2)如果发生任何更新需在60天内更新内容
3)产品清单负责人必须每年提供任何更新
五、FDA设施注册所需提交信息
1. 负责人名称、地址、邮箱、电话
2. FEI设施注册号
3. DUNS编码
4. 产品类别
5. 提交类型(初次、修订、续期)
6. 与注册相关的个人的联系信息
7.美代信息
8.产品Brand Name
六、产品注册所需提交信息
1. 化妆品成份清单(包括香料或成份通用名称)
2. 标签
3. 产品网页链接
4. 化妆品是否仅供专业人士使用
5. 产品清单编号
6. 提交类型(初始、更新或续订)
7. 独特成分标识符(UNIIS)
8. 业务类型(即制造商、包装商或分销商)
9. 与产品列明相关的个人联系信息
10.母公司名称(如有)
七、问答了解更多!
1.如果我的产品既是药品又是化妆品怎么办?
答:同时也是药物的化妆品不受FD&C法案第607条规定的要求进行设施和产品注册,按照药品注册。
2. 想FDA化妆品提交注册是否收取费用?
答:不收取注册费用。
3. 必须什么时间完成设施和产品注册?
答:1. 工厂注册
① 2022年12月29日之前就参与了化妆品生产或加工的企业,必须在2023年12月29日之前完成注册。
② 2022年12月29日之后首次参与化妆品生产或加工的企业,必须在首次从事此类活动后的60天内或2024年2月27日之前(以较晚者为准)完成注册。
2. 产品列名
① 2022年12月29日之前就上市的化妆品,必须在2023年12月29日之前完成列名。
② 2022年12月29日之后首次上市的化妆品,必须在首次上市后的120天内或2024年4月28日之前(以较晚者为准)完成注册。
4.化妆品设施注册和化妆品上市提交的信息可用于公开披露?
产品上市编号将不可公开披露,但化妆品工厂等信息将会被披露。
【华夏佳美】一站式跨境服务平台,专注医疗器械注册申请、GMP体系建立辅导、美国FDA510k、QSR820体系建立、欧盟CE认证、MDR、IVDR技术文件编写、ISO13485体系技术指导等提供全球医疗器械合规咨询服务,详情请咨询在线客服或官方电话:400-069-8010。
